Clinical Pipeline
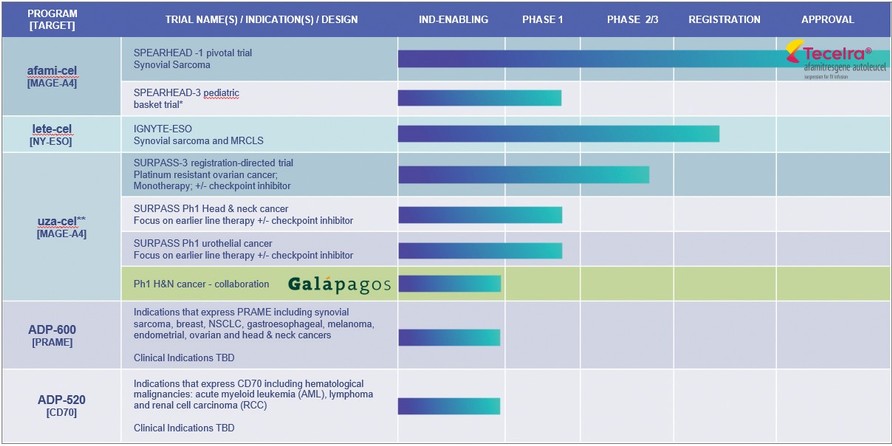
*Synovial sarcoma, Malignant Peripheral Nerve Sheath Tumor (MPNST), Neuroblastoma, Osteosarcoma, Temporary suspension of enrolment as per protocol in SPEARHEAD-3 trial
**uzatresgene autoleucel, formerly ADP-A2M4CD8; SURPASS Ph 1 no longer enrolling. Adaptimmune and Galapagos to conduct a clinical proof-of- concept trial to evaluate the safety and efficacy of uza-cel produced on Galapagos’ decentralized manufacturing platform in patients with
head & neck cancer
We have clinical trials ongoing in certain indications in which the MAGE-A4 antigen is expressed.
| ● | SPEARHEAD Trials with afami-cel. The SPEARHEAD trial is ongoing in the EU for treatment of patients with synovial sarcoma. A pediatric trial is ongoing in the US in tumors expressing the MAGE-A4 antigen, enrolment in this trial has been temporarily suspended as per protocol. |
| ● | SURPASS-3 Phase 2 Trial with uza-cel. A Phase 2 trial for people with platinum resistant ovarian cancer is ongoing. We have received Regenerative Medicine Advanced Therapy (“RMAT”) designation for uza-cel for the treatment of this indication from the FDA. The Phase 2 trial evaluates ADP-A2M4CD8 as both monotherapy and in combination with a checkpoint inhibitor, nivolumab, in ovarian cancer. The trial is open in the U.S., Canada, Spain, the U.K. and France. |
Our ADP-A2AFP Phase 1 trial, SURPASS-2 trial, gavo-cel and TC-510 trials have closed to enrollment. Screening for the SURPASS phase 1 trial has ceased and enrollment will close shortly.
Pre-clinical Pipeline
Our aim is to utilize the insights we obtain from our clinical trials and translational sciences work to improve the efficacy of our existing products and approaches; and to increase the scope of our cell therapies and ability to treat an increasing number of patients. We are currently focusing our preclinical pipeline on the development of T-cell therapies directed to PRAME (ADP-600) and CD70 (ADP-520) and on our allogeneic cell therapy platform.
| ● | PRAME is highly expressed across a broad range of solid tumors including ovarian, endometrial, lung and breast cancers. We are developing TCR T-cells directed to PRAME, with the initial candidate (ADP-600) currently in preclinical testing and next-generation candidates being developed over the longer term. |
| ● | The CD70 program targets the CD70 antigen which is expressed across a range of hematological malignancies (acute |
26