For all of our autologous cell therapies patient T-cells are extracted and are then engineered to generate the end cell therapy whether this is through engineering of the TCR itself or through the addition of another agent which enhances the efficacy of the TCR or T-cell. The nature of the engineering impacts the type of cell therapy product generated. The engineered T-cells are then expanded and infused back into the patient. When these T-cells encounter a recognized peptide or protein within the patient’s body, they multiply and initiate the destruction of the targeted cancer cells.
Adaptimmune has two receptor platforms, “TCRs” and “TRuCs”, which target different classes of antigen. Engineered TCRs target peptides from intracellular proteins that are naturally processed into peptide fragments and presented on the cell surface by HLA, whereas TRuCs bind to protein targets that are expressed on the cell surface, similar to the way in which Chimeric Antigen Receptor or “CAR” T-cells act. Following identification of a suitable target protein that is expected to have a safe expression profile, we tailor our approach depending on the extracellular or intracellular location of the target.
For intracellular target proteins we identify potential immunogenic peptides that are processed and presented by specific HLA types and then identify TCRs that are capable of binding to that specific peptide/HLA complex. We engineer and optimize those identified receptors to enhance their ability to recognize and bind to the cancer targets, thereby enabling a highly targeted immunotherapy which complements a patient’s immune system. The optimized TCR for the cell therapy then undergoes extensive preclinical safety testing prior to administration to patients who express the right protein target and HLA type. The majority of our products, current clinical candidates and most of our pre-clinical candidates target intracellular antigens presented by HLA-A2.
TRuCs use an antibody binding domain coupled to a CD3 subunit. The antibody portion binds to the target protein on the cancer cell and then the bound complex signals through a CD3/TCR complex using the natural signaling pathway of T cells. The physiological signaling method of TRuCs is distinct from CAR T-cell signaling where multiple signaling units are combined in a single protein. Natural TCRs are sensitive to much lower antigen density than CARs. Our ADP-520 preclinical program uses a TRuC directed to the CD70 antigen. There is no HLA restriction with this product meaning that all patients with tumors expressing CD70 could be eligible.
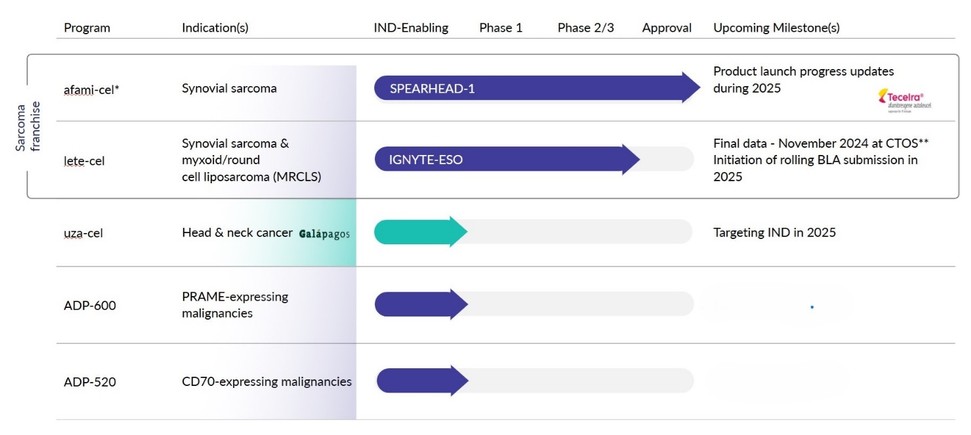
Our programs
TECELRA
In August 2024 we received accelerated approval from the FDA for TECELRA (famitresgene autoleucel or afami-cel) for adults with unresectable/metastatic synovial sarcoma who have received prior chemotherapy, are HLA-A*02 eligible, and whose tumor expresses MAGE-A4 as determined by FDA-approved diagnostics.
Afami-cel, afamitresgene autoleucel; HLA, human leukocyte antigen; MAGE-A4, melanoma-associated antigen A4; TCR, T-cell receptor.
Afami-cel consists of autologous T-cells engineered to express an affinity enhanced TCR targeting a MAGE-A4 peptide presented on cancer cells by certain human leukocyte antigens (HLAs).
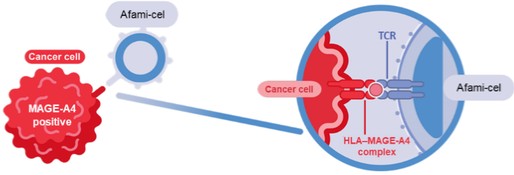
Afami-cel, afamitresgene autoleucel; HLA, human leukocyte antigen; MAGE-A4, melanoma-associated antigen A4; TCR, T-cell receptor.
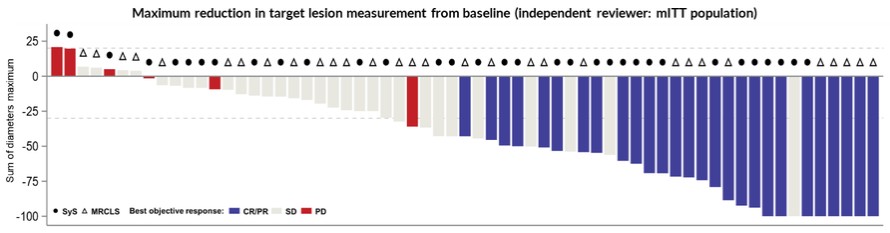
The approval of TECELRA was based on results from the SPEARHEAD-1 (Cohort 1) trial. In the primary analysis from the SPEARHEAD-1 trial, the pivotal trial for afami-cel, 52 patients were treated in cohort 1 of the trial. 44 synovial sarcoma patients and 8 myxoid liposarcoma patients. The overall response rate in the trial was 37% (19 out of
4